The second instalment in the ‘Ion Channels in Drug Discovery’ webinar series was presented by Professor Stephen Tucker from the Department of Physics, University of Oxford. Stephen delivered a presentation titled:
“Defective X-gating caused by de novo mutations in the TASK-1 K2P channel (KCNK3) underlies a developmental disorder with sleep apnoea”.
The work presented by Stephen is available as a pre-print in MedRxiv. The work was supported by Bayer who provided the TASK-1 blockers which were used in the study and are currently being advanced through clinical trials.
The origin of leak conductance
Stephen has spent much of his career working on the two-pore domain (K2P) and inward rectifier (Kir) families of potassium channels. K2P channels produce background leak currents that contribute to regulating the resting membrane potential. The first reported mammalian K2P channel, TWIK-1 (Tandem of pore domains in Weak Inward rectifying K+ channel 1, often referred to as KCNK1 or K2P1) was identified over 25 years ago and is now known to be one of 15 members in the human genome. These channels can be highly regulatable and finely tuneable; many exhibit polymodal regulation by diverse stimuli and cell signalling pathways, including GPCRs.
When K2P channels go wrong
K2P channels are expressed in a wide range of different tissue types where they have important roles in controlling physiological processes. Therefore, it is not surprising that variants that affect the function of K2P channels can result in a wide variety of disorders, which range from migraine to pulmonary arterial hypertension. The membrane topology of K2P channels differs significantly from ‘classical’ potassium channels, as each K2P channel subunit contains two pore forming domains and functional channels are composed of two subunits.
Disease causing mutations within the TASK channel family
TASK channels are a subset of the K2P channel family, which are expressed in many excitable and non-excitable tissues, including the pancreas, brain, lung, heart and kidney. Loss-of-function mutations in TASK-3 (KCNK9) can lead to a condition known as Birk Barel syndrome, an inherited condition characterised by intellectual disability, hyperactivity and unusual facial features. Most cases result from a mutation in the M4 helix, which produces almost complete loss-of-function. The condition demonstrates dominant inheritance with paternal imprinting. Furthermore, loss-of-function mutations in TASK-1 (KCNK3), (either homo- or heterozygous) have been linked to the pathophysiology of pulmonary arterial hypertension; a rare, progressive disorder.
What is the link with sleep apnoea?
TASK-1 (KCNK3) is also implicated in sleep apnoea, which is a disorder where breathing repeatedly stops during sleep. Sleep apnoea affects up to a billion people worldwide. and there are two main types:
TASK-1 channels are expressed in the chemosensitive regions of the brain that are involved in the control of breathing. Research showing the involvement of TASK-1 in sleep apnoea led Bayer to develop highly potent, highly selective inhibitors of TASK-1 channels that are currently in clinical development (phase 2).
A new channelopathy associated with TASK-1 (KCNK3)
The link between sleep apnoea and TASK-1 came from a recent publication based on the results of a large study of 31,000 parent-offspring trios. During the study, 28 novel genes were identified, including KCNK3 (TASK-1), that were associated with neurodevelopmental disorders such as developmental delay. A total of 9 probands, each heterozygous for one of six separate de novo missense mutations in KCNK3 were identified. Patients shared a common phenotype consisting of developmental delay with various limb abnormalities. Additionally, they also suffered from sleep apnoea that necessitated the use of nocturnal oxygen. The authors of the study termed the disorder, Developmental Delay with Sleep Apnoea (DDSA). Due to his expertise with TASK-1 channels, Stephen was contacted by the study leader to help characterise the mutant TASK-1 channels.
Obtaining structural information about the TASK1 channel
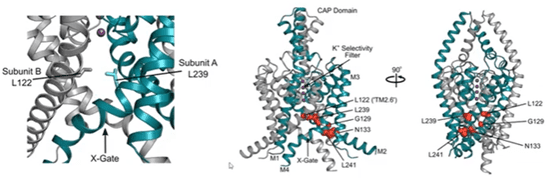
Stephen explained how his team obtained crystal structures of the TASK-1 channel (published in Nature in 2020). Interestingly, they identified that TASK-1 contains a lower gate, which they designated as an ‘X-gate’, which is created by interaction of the two crossed C-terminal M4 transmembrane helices at the vestibule entrance. This region is known as the halothane response element (HRE) and is important for TASK channel gating. The structures were co-crystalised with the Bayer inhibitors and the drug binding site was found to sit just below the selectivity filter. The six most prevalent mutations identified in DDSA are clustered around the X-gate.
Probing the functional characteristics of the channel using electrophysiology
Using the voltage clamp technique, it was observed that the mutant TASK-1 channels displayed a prominent gain-of-function phenotype, where dramatic increases in current amplitude were identified. The gain in function occurred irrespective of whether one or both of the channel subunits contained a mutation.
Single channel recordings performed from channels expressed in Xenopus oocytes, demonstrated a higher open probability for the TASK-1 mutants. N133S (TM2 mutation) and L239P (M4 mutation), were associated with a ten-fold increase in single channel open probability, but with no change in conductance. These mutations appear to be involved in interfering with interactions that stabilise the X-gate in its closed state. For example, N133 generates a backbone hydrogen bond with the M4 helix. When this residue was mutated, the H bond was disrupted, which led to a gain-in-channel function.. The group confirmed this result using a molecular dynamics simulation of the TASK-1 structure; wild type versus N133S and L239P. The two mutations were found to promote opening of the X-gate.
What regulates the X-gate?
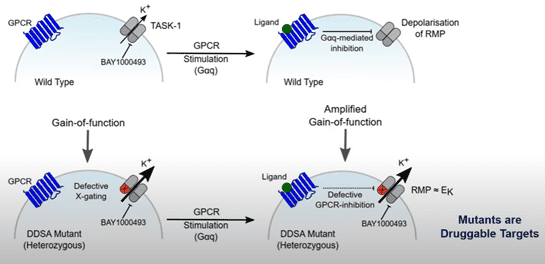
Previous studies which were performed almost 20 years ago, showed that mutating the X-gate renders TASK-1 resistant to GPCR mediated inhibition. Interestingly, all of the TASK-1 mutants associated with DDSA were resistant to GPCR inhibition, which may underlie what is happening with the L239P mutation. It was also found that the mutant channel, N133S, is not inhibited by either diacylglycerol or anandamide.
Can the mutant channels still be pharmacologically modulated?
A critical question regarding the mutant TASK-1 channels associated with DDSA, is whether they can be modulated by inhibitors of wildtype TASK-1. Bayer’s TASK-1 blocker, BAY 2253651, which is being developed for the treatment of obstructive sleep apnoea, inhibits TASK-1 with a sub nanomolar IC50. Fortunately, electrophysiological assessments identified that the mutations associated with DDSA are still sensitive to BAY 2253651; the compound is capable of significantly inhibiting the mutant channels.
Possible genotype/phenotype correlation
Mutations located in the M4 region produce the smallest increase in whole cell current amplitudes; patients with mutations in the M4 region presented with less severe phenotypes in general, exhibiting fewer limb abnormalities and less severe developmental delay. It will become clearer, with further research, whether this observation is consistent with other mutations that are identified in the M4 region. However, further work is required to further characterise the mutant channels, as these experiments were performed using Xenopus oocytes, rather than mammalian cells, and do not take into account factors such as hetero-multimerization with TASK-3 or coupling to different GPCRs.
Summary
It will be very interesting to see the final outcome of the Bayer clinical trials.
The second instalment in the ‘Ion Channels in Drug Discovery’ webinar series was presented by Professor Stephen Tucker from the Department of Physics, University of Oxford. Stephen delivered a presentation titled:
“Defective X-gating caused by de novo mutations in the TASK-1 K2P channel (KCNK3) underlies a developmental disorder with sleep apnoea”.
The work presented by Stephen is available as a pre-print in MedRxiv. The work was supported by Bayer who provided the TASK-1 blockers which were used in the study and are currently being advanced through clinical trials.
The origin of leak conductance
Stephen has spent much of his career working on the two-pore domain (K2P) and inward rectifier (Kir) families of potassium channels. K2P channels produce background leak currents that contribute to regulating the resting membrane potential. The first reported mammalian K2P channel, TWIK-1 (Tandem of pore domains in Weak Inward rectifying K+ channel 1, often referred to as KCNK1 or K2P1) was identified over 25 years ago and is now known to be one of 15 members in the human genome. These channels can be highly regulatable and finely tuneable; many exhibit polymodal regulation by diverse stimuli and cell signalling pathways, including GPCRs.
When K2P channels go wrong
K2P channels are expressed in a wide range of different tissue types where they have important roles in controlling physiological processes. Therefore, it is not surprising that variants that affect the function of K2P channels can result in a wide variety of disorders, which range from migraine to pulmonary arterial hypertension. The membrane topology of K2P channels differs significantly from ‘classical’ potassium channels, as each K2P channel subunit contains two pore forming domains and functional channels are composed of two subunits.
Disease causing mutations within the TASK channel family
TASK channels are a subset of the K2P channel family, which are expressed in many excitable and non-excitable tissues, including the pancreas, brain, lung, heart and kidney. Loss-of-function mutations in TASK-3 (KCNK9) can lead to a condition known as Birk Barel syndrome, an inherited condition characterised by intellectual disability, hyperactivity and unusual facial features. Most cases result from a mutation in the M4 helix, which produces almost complete loss-of-function. The condition demonstrates dominant inheritance with paternal imprinting. Furthermore, loss-of-function mutations in TASK-1 (KCNK3), (either homo- or heterozygous) have been linked to the pathophysiology of pulmonary arterial hypertension; a rare, progressive disorder.
What is the link with sleep apnoea?
TASK-1 (KCNK3) is also implicated in sleep apnoea, which is a disorder where breathing repeatedly stops during sleep. Sleep apnoea affects up to a billion people worldwide. and there are two main types:
TASK-1 channels are expressed in the chemosensitive regions of the brain that are involved in the control of breathing. Research showing the involvement of TASK-1 in sleep apnoea led Bayer to develop highly potent, highly selective inhibitors of TASK-1 channels that are currently in clinical development (phase 2).
A new channelopathy associated with TASK-1 (KCNK3)
The link between sleep apnoea and TASK-1 came from a recent publication based on the results of a large study of 31,000 parent-offspring trios. During the study, 28 novel genes were identified, including KCNK3 (TASK-1), that were associated with neurodevelopmental disorders such as developmental delay. A total of 9 probands, each heterozygous for one of six separate de novo missense mutations in KCNK3 were identified. Patients shared a common phenotype consisting of developmental delay with various limb abnormalities. Additionally, they also suffered from sleep apnoea that necessitated the use of nocturnal oxygen. The authors of the study termed the disorder, Developmental Delay with Sleep Apnoea (DDSA). Due to his expertise with TASK-1 channels, Stephen was contacted by the study leader to help characterise the mutant TASK-1 channels.
Obtaining structural information about the TASK1 channel
Stephen explained how his team obtained crystal structures of the TASK-1 channel (published in Nature in 2020). Interestingly, they identified that TASK-1 contains a lower gate, which they designated as an ‘X-gate’, which is created by interaction of the two crossed C-terminal M4 transmembrane helices at the vestibule entrance. This region is known as the halothane response element (HRE) and is important for TASK channel gating. The structures were co-crystalised with the Bayer inhibitors and the drug binding site was found to sit just below the selectivity filter. The six most prevalent mutations identified in DDSA are clustered around the X-gate.
Probing the functional characteristics of the channel using electrophysiology
Using the voltage clamp technique, it was observed that the mutant TASK-1 channels displayed a prominent gain-of-function phenotype, where dramatic increases in current amplitude were identified. The gain in function occurred irrespective of whether one or both of the channel subunits contained a mutation.
Single channel recordings performed from channels expressed in Xenopus oocytes, demonstrated a higher open probability for the TASK-1 mutants. N133S (TM2 mutation) and L239P (M4 mutation), were associated with a ten-fold increase in single channel open probability, but with no change in conductance. These mutations appear to be involved in interfering with interactions that stabilise the X-gate in its closed state. For example, N133 generates a backbone hydrogen bond with the M4 helix. When this residue was mutated, the H bond was disrupted, which led to a gain-in-channel function.. The group confirmed this result using a molecular dynamics simulation of the TASK-1 structure; wild type versus N133S and L239P. The two mutations were found to promote opening of the X-gate.
What regulates the X-gate?
Previous studies which were performed almost 20 years ago, showed that mutating the X-gate renders TASK-1 resistant to GPCR mediated inhibition. Interestingly, all of the TASK-1 mutants associated with DDSA were resistant to GPCR inhibition, which may underlie what is happening with the L239P mutation. It was also found that the mutant channel, N133S, is not inhibited by either diacylglycerol or anandamide.
Can the mutant channels still be pharmacologically modulated?
A critical question regarding the mutant TASK-1 channels associated with DDSA, is whether they can be modulated by inhibitors of wildtype TASK-1. Bayer’s TASK-1 blocker, BAY 2253651, which is being developed for the treatment of obstructive sleep apnoea, inhibits TASK-1 with a sub nanomolar IC50. Fortunately, electrophysiological assessments identified that the mutations associated with DDSA are still sensitive to BAY 2253651; the compound is capable of significantly inhibiting the mutant channels.
Possible genotype/phenotype correlation
Mutations located in the M4 region produce the smallest increase in whole cell current amplitudes; patients with mutations in the M4 region presented with less severe phenotypes in general, exhibiting fewer limb abnormalities and less severe developmental delay. It will become clearer, with further research, whether this observation is consistent with other mutations that are identified in the M4 region. However, further work is required to further characterise the mutant channels, as these experiments were performed using Xenopus oocytes, rather than mammalian cells, and do not take into account factors such as hetero-multimerization with TASK-3 or coupling to different GPCRs.
Summary
It will be very interesting to see the final outcome of the Bayer clinical trials.
The second instalment in the ‘Ion Channels in Drug Discovery’ webinar series was presented by Professor Stephen Tucker from the Department of Physics, University of Oxford. Stephen delivered a presentation titled:
“Defective X-gating caused by de novo mutations in the TASK-1 K2P channel (KCNK3) underlies a developmental disorder with sleep apnoea”.
The work presented by Stephen is available as a pre-print in MedRxiv. The work was supported by Bayer who provided the TASK-1 blockers which were used in the study and are currently being advanced through clinical trials.
The origin of leak conductance
Stephen has spent much of his career working on the two-pore domain (K2P) and inward rectifier (Kir) families of potassium channels. K2P channels produce background leak currents that contribute to regulating the resting membrane potential. The first reported mammalian K2P channel, TWIK-1 (Tandem of pore domains in Weak Inward rectifying K+ channel 1, often referred to as KCNK1 or K2P1) was identified over 25 years ago and is now known to be one of 15 members in the human genome. These channels can be highly regulatable and finely tuneable; many exhibit polymodal regulation by diverse stimuli and cell signalling pathways, including GPCRs.
When K2P channels go wrong
K2P channels are expressed in a wide range of different tissue types where they have important roles in controlling physiological processes. Therefore, it is not surprising that variants that affect the function of K2P channels can result in a wide variety of disorders, which range from migraine to pulmonary arterial hypertension. The membrane topology of K2P channels differs significantly from ‘classical’ potassium channels, as each K2P channel subunit contains two pore forming domains and functional channels are composed of two subunits.
Disease causing mutations within the TASK channel family
TASK channels are a subset of the K2P channel family, which are expressed in many excitable and non-excitable tissues, including the pancreas, brain, lung, heart and kidney. Loss-of-function mutations in TASK-3 (KCNK9) can lead to a condition known as Birk Barel syndrome, an inherited condition characterised by intellectual disability, hyperactivity and unusual facial features. Most cases result from a mutation in the M4 helix, which produces almost complete loss-of-function. The condition demonstrates dominant inheritance with paternal imprinting. Furthermore, loss-of-function mutations in TASK-1 (KCNK3), (either homo- or heterozygous) have been linked to the pathophysiology of pulmonary arterial hypertension; a rare, progressive disorder.
What is the link with sleep apnoea?
TASK-1 (KCNK3) is also implicated in sleep apnoea, which is a disorder where breathing repeatedly stops during sleep. Sleep apnoea affects up to a billion people worldwide. and there are two main types:
TASK-1 channels are expressed in the chemosensitive regions of the brain that are involved in the control of breathing. Research showing the involvement of TASK-1 in sleep apnoea led Bayer to develop highly potent, highly selective inhibitors of TASK-1 channels that are currently in clinical development (phase 2).
A new channelopathy associated with TASK-1 (KCNK3)
The link between sleep apnoea and TASK-1 came from a recent publication based on the results of a large study of 31,000 parent-offspring trios. During the study, 28 novel genes were identified, including KCNK3 (TASK-1), that were associated with neurodevelopmental disorders such as developmental delay. A total of 9 probands, each heterozygous for one of six separate de novo missense mutations in KCNK3 were identified. Patients shared a common phenotype consisting of developmental delay with various limb abnormalities. Additionally, they also suffered from sleep apnoea that necessitated the use of nocturnal oxygen. The authors of the study termed the disorder, Developmental Delay with Sleep Apnoea (DDSA). Due to his expertise with TASK-1 channels, Stephen was contacted by the study leader to help characterise the mutant TASK-1 channels.
Obtaining structural information about the TASK1 channel
Stephen explained how his team obtained crystal structures of the TASK-1 channel (published in Nature in 2020). Interestingly, they identified that TASK-1 contains a lower gate, which they designated as an ‘X-gate’, which is created by interaction of the two crossed C-terminal M4 transmembrane helices at the vestibule entrance. This region is known as the halothane response element (HRE) and is important for TASK channel gating. The structures were co-crystalised with the Bayer inhibitors and the drug binding site was found to sit just below the selectivity filter. The six most prevalent mutations identified in DDSA are clustered around the X-gate.
Probing the functional characteristics of the channel using electrophysiology
Using the voltage clamp technique, it was observed that the mutant TASK-1 channels displayed a prominent gain-of-function phenotype, where dramatic increases in current amplitude were identified. The gain in function occurred irrespective of whether one or both of the channel subunits contained a mutation.
Single channel recordings performed from channels expressed in Xenopus oocytes, demonstrated a higher open probability for the TASK-1 mutants. N133S (TM2 mutation) and L239P (M4 mutation), were associated with a ten-fold increase in single channel open probability, but with no change in conductance. These mutations appear to be involved in interfering with interactions that stabilise the X-gate in its closed state. For example, N133 generates a backbone hydrogen bond with the M4 helix. When this residue was mutated, the H bond was disrupted, which led to a gain-in-channel function.. The group confirmed this result using a molecular dynamics simulation of the TASK-1 structure; wild type versus N133S and L239P. The two mutations were found to promote opening of the X-gate.
What regulates the X-gate?
Previous studies which were performed almost 20 years ago, showed that mutating the X-gate renders TASK-1 resistant to GPCR mediated inhibition. Interestingly, all of the TASK-1 mutants associated with DDSA were resistant to GPCR inhibition, which may underlie what is happening with the L239P mutation. It was also found that the mutant channel, N133S, is not inhibited by either diacylglycerol or anandamide.
Can the mutant channels still be pharmacologically modulated?
A critical question regarding the mutant TASK-1 channels associated with DDSA, is whether they can be modulated by inhibitors of wildtype TASK-1. Bayer’s TASK-1 blocker, BAY 2253651, which is being developed for the treatment of obstructive sleep apnoea, inhibits TASK-1 with a sub nanomolar IC50. Fortunately, electrophysiological assessments identified that the mutations associated with DDSA are still sensitive to BAY 2253651; the compound is capable of significantly inhibiting the mutant channels.
Possible genotype/phenotype correlation
Mutations located in the M4 region produce the smallest increase in whole cell current amplitudes; patients with mutations in the M4 region presented with less severe phenotypes in general, exhibiting fewer limb abnormalities and less severe developmental delay. It will become clearer, with further research, whether this observation is consistent with other mutations that are identified in the M4 region. However, further work is required to further characterise the mutant channels, as these experiments were performed using Xenopus oocytes, rather than mammalian cells, and do not take into account factors such as hetero-multimerization with TASK-3 or coupling to different GPCRs.
Summary
It will be very interesting to see the final outcome of the Bayer clinical trials.