Today, we mark Rare Disease Day by highlighting a selection of the most detrimental ion channel mutations which can result in a plethora of rare diseases, often involving complex neurological, cardiac, and muscular manifestations.
The ion channels implicated in such conditions are of several types: voltage-gated sodium channels, chloride channels, calcium channels, and potassium channels. These channels mediate vital biological processes, from the generation of electrical signals in nerves to maintaining the balance of fluids in organs such as the kidneys and lungs.
One of the most studied ion channels is the voltage-gated sodium channel, particularly SCN1A (NaV1.1), which is associated with Dravet Syndrome, a severe form of epilepsy that typically manifests in infancy. Mutations in SCN1A disrupt the normal flow of sodium ions into neurons, impairing their ability to communicate effectively and leading to the development of uncontrolled seizures, cognitive impairments, and motor dysfunction1.
Another well-known channel implicated in rare diseases is the CFTR chloride channel. Mutations in the CFTR gene lead to cystic fibrosis, a genetic disorder that primarily affects the lungs and digestive system, causing thick mucus build-up and chronic infections that can significantly shorten life expectancy2.
One example of a calcium channelopathy involves the CACNA1A (CaV2.1) gene, which encodes a subunit of the voltage-gated calcium channel involved in muscle contraction and neurotransmitter release. Mutations in this gene are linked to a variety of conditions, including familial hemiplegic migraine and episodic ataxia, both of which can result in debilitating episodes of vertigo, ataxia, and severe headaches3.
Potassium channels play a pivotal role in regulating neuronal excitability, muscle contraction, and heart rhythms and amongst the most significant potassium channels linked to rare diseases are KCNQ2 (KV7.2) and KCNC1 (KV3.1).
KCNC1 encodes for a voltage-gated potassium channel found in neurons. Mutations in KCNC1 are associated with a range of neurological disorders, including myoclonic epilepsy, ataxia, and movement disorders4. This highlights the critical role that potassium channels play in neuronal function, where disruptions in the delicate balance of potassium ion flow can have profound consequences. KCNC1 Foundation, in collaboration with Perlara, commissioned Metrion Biosciences to undertake drug repurposing studies to identify modulators of the channel as therapies for a two-year-old with a de novo KCNC1 mutation (Figure 1). Read our case study (Multi-assay High-throughput Drug Repurposing Screen: KCNC1 Case Study) and watch our webinar (Recent Progress Towards a Potential Treatment for KCNC-1 Related Disorders) to find out how this key study unfolded.
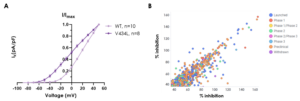
Figure 1. Biophysical characterisation of the V434L-variant of KV3.1 channel (KCNC1) and a high-throughput drug repurposing screen. A Representative current density plots for voltage-dependent activation for wild-type (WT) and V434L-variant KV3.1 channels, recorded by manual patch-clamp in CHO-K1 cells. The clear leftward shift confirms V434L as a gain-of-function mutation. B Correlation of duplicate percentage inhibition data with compounds inhibiting the V434L-variant KV3.1 channel greater than 50% annotated with phase in drug development. The repurposing screen has been performed using thallium flux assays on the FLIPR® Penta (Molecular Devices) platform.
Similarly, KCNQ2 mutations are most notably associated with cases of early-onset epileptic encephalopathy, as this gene encodes a potassium channel critical for neuronal excitability. As a result, individuals with KCNQ2 mutations often experience severe developmental delays, recurrent seizures, and in some cases, intellectual disability, making it a key factor in certain rare and challenging neurological diseases5.
Brugada syndrome highlights the importance of ion channels in maintaining the electrical stability of the heart. This is a rare but significant condition caused by mutations in the SCN5A gene, which encodes a sodium channel critical for heart electrical signalling. These mutations can lead to life-threatening arrhythmias, syncope, and sudden cardiac arrest. Several other diseases are associated with SCN5A mutations, such as the Long QT syndrome and Lev (Lenègre) syndrome6.
Bartter Syndrome is a rare genetic condition caused by mutations in ion channels such as SLC12A17, KCNJ18, and CLCNKB9 that are critical for salt reabsorption in the kidneys. The condition leads to electrolyte imbalances, including hypokalaemia (low potassium levels), and can cause dehydration, muscle weakness, and metabolic alkalosis.
Although not directly linked to ion channel dysfunction, Duchenne Muscular Dystrophy (DMD) is associated with impaired ion channel activity. In DMD, a defect in the dystrophin protein affects the ability of muscle cells to function properly, causing progressive muscle weakness and loss of muscle mass. While DMD is primarily caused by mutations in the dystrophin gene, diverse types of ion channel dysfunction also play a role in the pathophysiology of muscle excitability and contraction, with emerging therapeutic implications involving ion channel modulation10.
Metrion offers the unique combination of highly experienced ion channel scientists and cutting-edge ion channel screening platforms across multiple disciplines, enabling you to study specific ion channels and their involvement in rare diseases.
By generating custom cell lines carrying specific mutations, developing customised high-throughput screening assays and accessing compound libraries, we can help you identify therapies that could restore ion channel function and improve patient outcomes. This is crucial for advancing personalised medicine and offering hope to individuals affected by these rare and devastating conditions.
Contact us to discover how we can help drive your project forward.
Today, we mark Rare Disease Day by highlighting a selection of the most detrimental ion channel mutations which can result in a plethora of rare diseases, often involving complex neurological, cardiac, and muscular manifestations.
The ion channels implicated in such conditions are of several types: voltage-gated sodium channels, chloride channels, calcium channels, and potassium channels. These channels mediate vital biological processes, from the generation of electrical signals in nerves to maintaining the balance of fluids in organs such as the kidneys and lungs.
One of the most studied ion channels is the voltage-gated sodium channel, particularly SCN1A (NaV1.1), which is associated with Dravet Syndrome, a severe form of epilepsy that typically manifests in infancy. Mutations in SCN1A disrupt the normal flow of sodium ions into neurons, impairing their ability to communicate effectively and leading to the development of uncontrolled seizures, cognitive impairments, and motor dysfunction1.
Another well-known channel implicated in rare diseases is the CFTR chloride channel. Mutations in the CFTR gene lead to cystic fibrosis, a genetic disorder that primarily affects the lungs and digestive system, causing thick mucus build-up and chronic infections that can significantly shorten life expectancy2.
One example of a calcium channelopathy involves the CACNA1A (CaV2.1) gene, which encodes a subunit of the voltage-gated calcium channel involved in muscle contraction and neurotransmitter release. Mutations in this gene are linked to a variety of conditions, including familial hemiplegic migraine and episodic ataxia, both of which can result in debilitating episodes of vertigo, ataxia, and severe headaches3.
Potassium channels play a pivotal role in regulating neuronal excitability, muscle contraction, and heart rhythms and amongst the most significant potassium channels linked to rare diseases are KCNQ2 (KV7.2) and KCNC1 (KV3.1).
KCNC1 encodes for a voltage-gated potassium channel found in neurons. Mutations in KCNC1 are associated with a range of neurological disorders, including myoclonic epilepsy, ataxia, and movement disorders4. This highlights the critical role that potassium channels play in neuronal function, where disruptions in the delicate balance of potassium ion flow can have profound consequences. KCNC1 Foundation, in collaboration with Perlara, commissioned Metrion Biosciences to undertake drug repurposing studies to identify modulators of the channel as therapies for a two-year-old with a de novo KCNC1 mutation (Figure 1). Read our case study (Multi-assay High-throughput Drug Repurposing Screen: KCNC1 Case Study) and watch our webinar (Recent Progress Towards a Potential Treatment for KCNC-1 Related Disorders) to find out how this key study unfolded.
Figure 1. Biophysical characterisation of the V434L-variant of KV3.1 channel (KCNC1) and a high-throughput drug repurposing screen. A Representative current density plots for voltage-dependent activation for wild-type (WT) and V434L-variant KV3.1 channels, recorded by manual patch-clamp in CHO-K1 cells. The clear leftward shift confirms V434L as a gain-of-function mutation. B Correlation of duplicate percentage inhibition data with compounds inhibiting the V434L-variant KV3.1 channel greater than 50% annotated with phase in drug development. The repurposing screen has been performed using thallium flux assays on the FLIPR® Penta (Molecular Devices) platform.
Similarly, KCNQ2 mutations are most notably associated with cases of early-onset epileptic encephalopathy, as this gene encodes a potassium channel critical for neuronal excitability. As a result, individuals with KCNQ2 mutations often experience severe developmental delays, recurrent seizures, and in some cases, intellectual disability, making it a key factor in certain rare and challenging neurological diseases5.
Brugada syndrome highlights the importance of ion channels in maintaining the electrical stability of the heart. This is a rare but significant condition caused by mutations in the SCN5A gene, which encodes a sodium channel critical for heart electrical signalling. These mutations can lead to life-threatening arrhythmias, syncope, and sudden cardiac arrest. Several other diseases are associated with SCN5A mutations, such as the Long QT syndrome and Lev (Lenègre) syndrome6.
Bartter Syndrome is a rare genetic condition caused by mutations in ion channels such as SLC12A17, KCNJ18, and CLCNKB9 that are critical for salt reabsorption in the kidneys. The condition leads to electrolyte imbalances, including hypokalaemia (low potassium levels), and can cause dehydration, muscle weakness, and metabolic alkalosis.
Although not directly linked to ion channel dysfunction, Duchenne Muscular Dystrophy (DMD) is associated with impaired ion channel activity. In DMD, a defect in the dystrophin protein affects the ability of muscle cells to function properly, causing progressive muscle weakness and loss of muscle mass. While DMD is primarily caused by mutations in the dystrophin gene, diverse types of ion channel dysfunction also play a role in the pathophysiology of muscle excitability and contraction, with emerging therapeutic implications involving ion channel modulation10.
Metrion offers the unique combination of highly experienced ion channel scientists and cutting-edge ion channel screening platforms across multiple disciplines, enabling you to study specific ion channels and their involvement in rare diseases.
By generating custom cell lines carrying specific mutations, developing customised high-throughput screening assays and accessing compound libraries, we can help you identify therapies that could restore ion channel function and improve patient outcomes. This is crucial for advancing personalised medicine and offering hope to individuals affected by these rare and devastating conditions.
Contact us to discover how we can help drive your project forward.
Today, we mark Rare Disease Day by highlighting a selection of the most detrimental ion channel mutations which can result in a plethora of rare diseases, often involving complex neurological, cardiac, and muscular manifestations.
The ion channels implicated in such conditions are of several types: voltage-gated sodium channels, chloride channels, calcium channels, and potassium channels. These channels mediate vital biological processes, from the generation of electrical signals in nerves to maintaining the balance of fluids in organs such as the kidneys and lungs.
One of the most studied ion channels is the voltage-gated sodium channel, particularly SCN1A (NaV1.1), which is associated with Dravet Syndrome, a severe form of epilepsy that typically manifests in infancy. Mutations in SCN1A disrupt the normal flow of sodium ions into neurons, impairing their ability to communicate effectively and leading to the development of uncontrolled seizures, cognitive impairments, and motor dysfunction1.
Another well-known channel implicated in rare diseases is the CFTR chloride channel. Mutations in the CFTR gene lead to cystic fibrosis, a genetic disorder that primarily affects the lungs and digestive system, causing thick mucus build-up and chronic infections that can significantly shorten life expectancy2.
One example of a calcium channelopathy involves the CACNA1A (CaV2.1) gene, which encodes a subunit of the voltage-gated calcium channel involved in muscle contraction and neurotransmitter release. Mutations in this gene are linked to a variety of conditions, including familial hemiplegic migraine and episodic ataxia, both of which can result in debilitating episodes of vertigo, ataxia, and severe headaches3.
Potassium channels play a pivotal role in regulating neuronal excitability, muscle contraction, and heart rhythms and amongst the most significant potassium channels linked to rare diseases are KCNQ2 (KV7.2) and KCNC1 (KV3.1).
KCNC1 encodes for a voltage-gated potassium channel found in neurons. Mutations in KCNC1 are associated with a range of neurological disorders, including myoclonic epilepsy, ataxia, and movement disorders4. This highlights the critical role that potassium channels play in neuronal function, where disruptions in the delicate balance of potassium ion flow can have profound consequences. KCNC1 Foundation, in collaboration with Perlara, commissioned Metrion Biosciences to undertake drug repurposing studies to identify modulators of the channel as therapies for a two-year-old with a de novo KCNC1 mutation (Figure 1). Read our case study (Multi-assay High-throughput Drug Repurposing Screen: KCNC1 Case Study) and watch our webinar (Recent Progress Towards a Potential Treatment for KCNC-1 Related Disorders) to find out how this key study unfolded.
Figure 1. Biophysical characterisation of the V434L-variant of KV3.1 channel (KCNC1) and a high-throughput drug repurposing screen. A Representative current density plots for voltage-dependent activation for wild-type (WT) and V434L-variant KV3.1 channels, recorded by manual patch-clamp in CHO-K1 cells. The clear leftward shift confirms V434L as a gain-of-function mutation. B Correlation of duplicate percentage inhibition data with compounds inhibiting the V434L-variant KV3.1 channel greater than 50% annotated with phase in drug development. The repurposing screen has been performed using thallium flux assays on the FLIPR® Penta (Molecular Devices) platform.
Similarly, KCNQ2 mutations are most notably associated with cases of early-onset epileptic encephalopathy, as this gene encodes a potassium channel critical for neuronal excitability. As a result, individuals with KCNQ2 mutations often experience severe developmental delays, recurrent seizures, and in some cases, intellectual disability, making it a key factor in certain rare and challenging neurological diseases5.
Brugada syndrome highlights the importance of ion channels in maintaining the electrical stability of the heart. This is a rare but significant condition caused by mutations in the SCN5A gene, which encodes a sodium channel critical for heart electrical signalling. These mutations can lead to life-threatening arrhythmias, syncope, and sudden cardiac arrest. Several other diseases are associated with SCN5A mutations, such as the Long QT syndrome and Lev (Lenègre) syndrome6.
Bartter Syndrome is a rare genetic condition caused by mutations in ion channels such as SLC12A17, KCNJ18, and CLCNKB9 that are critical for salt reabsorption in the kidneys. The condition leads to electrolyte imbalances, including hypokalaemia (low potassium levels), and can cause dehydration, muscle weakness, and metabolic alkalosis.
Although not directly linked to ion channel dysfunction, Duchenne Muscular Dystrophy (DMD) is associated with impaired ion channel activity. In DMD, a defect in the dystrophin protein affects the ability of muscle cells to function properly, causing progressive muscle weakness and loss of muscle mass. While DMD is primarily caused by mutations in the dystrophin gene, diverse types of ion channel dysfunction also play a role in the pathophysiology of muscle excitability and contraction, with emerging therapeutic implications involving ion channel modulation10.
Metrion offers the unique combination of highly experienced ion channel scientists and cutting-edge ion channel screening platforms across multiple disciplines, enabling you to study specific ion channels and their involvement in rare diseases.
By generating custom cell lines carrying specific mutations, developing customised high-throughput screening assays and accessing compound libraries, we can help you identify therapies that could restore ion channel function and improve patient outcomes. This is crucial for advancing personalised medicine and offering hope to individuals affected by these rare and devastating conditions.
Contact us to discover how we can help drive your project forward.